Identification of unstable CNG repeat loci in the human genome: a heuristic approach and implications for neurological disorders – Human Genome Variation
Identification of CNG repeats from the human reference genome sequence
Through genome-wide CNG repeat selection, we found a total of 15,069 loci (≥ 4 consecutive repeats) (Fig. 1). CNG repeats were abundant in the coding region and UTR. In general, CAG and CTG repeats were more abundant in different genomic regions (Table 1). We scored these replicate sites using ANNOVAR8 and further categorized the tandem repeats based on the length observed in the reference genome: Group 1, 4–6 repeats; Group 2, 7–9 repetitions; and Group 3, >9 replicates (Table 1).
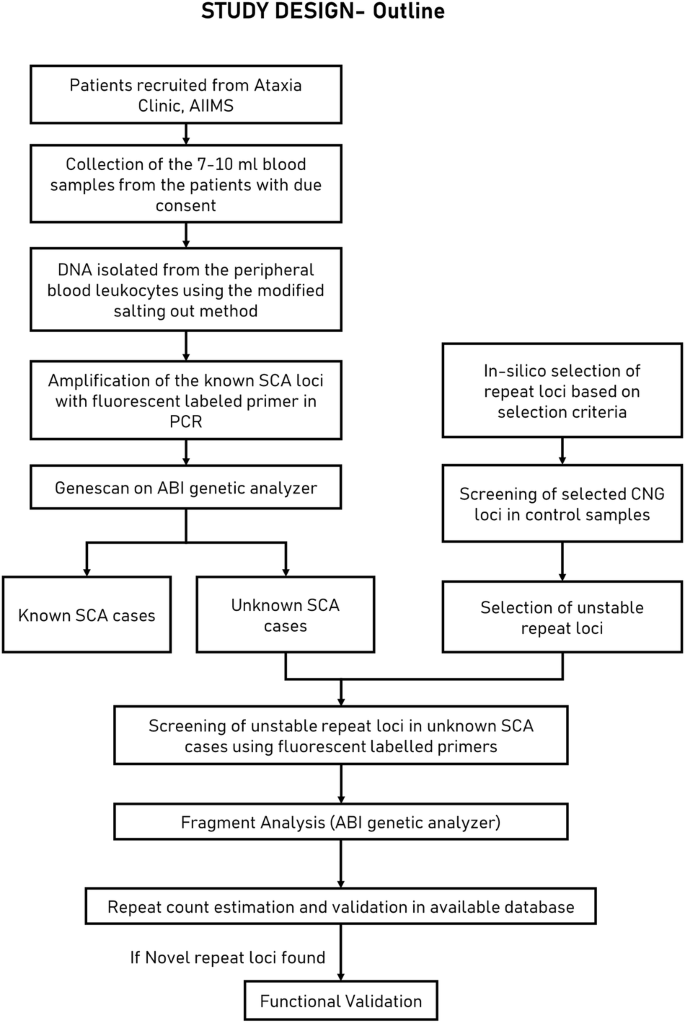
In silico, selected tandem repeats were assessed for their instability and further examined in the spinocerebellar ataxia disorder patient population.
Using a reductionist approach for further analysis, we selected 52 loci located in the CDS or UTR region with a tandem repeat length ≥10 ( Table 2 and Fig. 2 ). Repeats with more than 10 units are more prone to expansion events9 and cause a decrease in flap-1 endonuclease activity (FEN1) in Okazaki fragments10. Furthermore, most pathogenic trinucleotide repeat expansions were observed in the coding region or UTR, for example, in SCA1-SCA3 (CAG expansion in the coding region), SCA12 (CAG expansion in the 5′ UTR) and myotonic dystrophy (CTG extension in UTR 3).

Distribution of repeated categories within groups, showing percentage of repetitions per category, with colors representing different variables.
Genotyping of 52 CNG replicates in an Indian control population
By evaluating the length variability of 52 loci in control samples, 33 loci were found to be relatively stable (length variability of 1–6 repeat units) and 19 loci were more polymorphic in nature (length variability of 7–23 repeated units). These 19 other repeated variable sites (RAI1, UMAD1, GLS, HTR7P1, CNKSR2, MAML3, MED15, MLLT3, USF3, MEF2A, MIR205HG, NCOR2, RPL14, JPH3, MAB21L1, ANKUB1, ERF, GIPC1AND EP400) were further examined in our cohort of ataxia patients to identify any length variations that may be pathogenic ( Fig. 3 ).

Distribution of target repeats among control and patient samples.
of MAB21L1, ANKUB1AND GLS genes were highly polymorphic and had a wide range of repeat distributions in the population [modes of repeats (ranges): 13 (8–26), 15 (8–33), and 12 (6–29), respectively]. Genes ANKUB1 AND UMAD1 showed a large number of repeats (> 30 repeats) in both case and control groups. No significant difference in gross dilation range was observed between case and control examinations (Table 2).
Heterozygosity indices (HIs, which measure the number of heterozygotes in the population) of UMAD1, MAB21L1, ANKUB1, GLSAND RPL14 were greater than 0.7 in both cases and controls. On the other side, MLLT3 AND CNKSR2 were less polymorphic and had more homozygous repeats (HI ≤ 0.1) in both groups. Most target loci fell within the range of 0.3 to 0.7, with the exception of ERF, which had an HI of less than 0.25 in all samples.
Selection of unstable CNG repeats in the 1000 Genomes database
Since disease-associated tandem repeats tend to be more polymorphic in the general population, we investigated the polymorphic nature of these loci in the control population. Compared to the different 1000 Genomes control populations, the mode of repeats and variability in the GLS gene were greater in African and SAS populations (Table 3). MAB21L1 showed a greater range of recurrence in the EAS population. Although some of the other loci had a maximum of >20 repeat expansions, these loci were uniform or less variable within populations. MEF2A was highly variable, ranging from 2 to 16 repeats, but was uniform across the population. GIPC1 Recurrence variability was less common in the EUR population. ABOUT MED15 AND ERF, replicate data were available for very few patient samples among different populations. We could not find any short repeat data together for HTR7P1, RPL14, CNKSR2OR MLLT3 repeat locations. Our repeat data for GLS, ANKUB1, EP400, JPH3AND RAI1 loci showed a biallelic distribution, which is also observed in other major populations.
Interestingly, we observed variability in the recurrence intervals of USF3, MEF2A, JPH3, RAI1, ERF, MED15, MAML3AND UMAD1 compared to those of other world populations, but none of the differences were significant according to the Wilcoxon signed rank test (nonparametric test). Both our groups had relatively fewer repeats for the EP400 loci (Table 4). The possible reason for this difference is the use of different sequencing technologies; short-read sequencing was used for the 1000 Genomes Project data. While short-read sequencing has its advantages, it also has some inherent inefficiencies with respect to capturing long-range repeats and complex genomic regions.
Analysis of expression levels of genes containing unstable repeats
For all candidate genes, the large tissue gene expression of each gene was compared between different tissues using GTEx11. The analysis showed that CNKSR2, MAB21L1, USF3, RAI1, NCOR2, JPH3, MAML3, EP400AND GLS genes were significantly expressed in the brain, particularly in the cerebellum. All other genes except MIR205HG, also showed significant expression levels in the brain (Table 2). Since the pathogenesis of SCA is related to the brain, we excluded it MIR205HG from the shortlist of genes. Thus, we proposed the pathogenicity of the remaining 18 genes, which may indicate an ataxia phenotype.
#Identification #unstable #CNG #repeat #loci #human #genome #heuristic #approach #implications #neurological #disorders #Human #Genome #Variation
Image Source : www.nature.com